Advantages
- Detection and quantification of over 10,000 proteins and client specified PTM’s, from 10 to 100s of fluid and tissue samples.
- Ability to enrich for specific protein subsets, such as secreted proteins, phosphopeptides and other post translational modifications and membrane proteins.
- Due to the unbiased nature of the analysis, we report all proteins/PTM’s that are detectable, unlike antibody or aptamer/affimer based technologies where the data is biased to the make-up of the selected analysis panel.
- By using an upfront top-14 depletion stage, we can quantify significantly more lower abundant phosphoproteins in plasma and serum.
- Multiplexing multiple samples in a single TMT based mass spectrometry run significantly increases the sensitivity of protein detection.
- Compatibility with a wide variety of sample matrices, including tissue. plasma, serum, cell lysates, CSF, exosomes etc.
- Available for human, rodent, monkey, pig and other higher animal species.
- With TMT, multiple samples can be analysed on one mass spectrometric run. Using a common reference channel per plex we can quantify samples from 17 to 100+ sample cohorts in multiple mass spectrometric runs.
- Generally technique is more sensitive than the same assay performed with label free mass spectrometry.
- With TMT isobaric tags we can report as low as 30% changes in a particular protein in your samples vrs 3 to 4 fold with label free methods.
- Due to the unbiased nature of the analysis, we report see all proteins that are detectable. Data collection is unbiased.
- Analysis package includes full computational proteomic analysis and bioinformatics covering extensive data interpretation, pathway analysis and biological relevance.
| Application | Service | Description |
|---|---|---|
|
Routine Biomarker Assays Pathway Mapping Drug mode of action Cell Signalling |
SysQuant to quantify both the proteome and specified post translational modifications |
|
Post-translational modifications (PTM’s) are key regulators of cell state and control the localisation, function and stability of proteins. Co-ordinated changes in phosphorylation in particular, are master regulators of cell signalling and regulatory processes and is often dysregulated in disease. Assessing the phosphorylation status of proteins cannot be inferred from genomic or transcriptomic data, and requires highly specific localisation, seldom possible using antibodies. Consequently, the use of mass spectrometry for interrogation of the phosphoproteome has been an important tool in understanding disease biology and the mechanism of action of drugs.
Proteome Sciences has developed SysQuant - a TMTpro enabled workflow combining standard proteomic analysis with phospho (and other PTM) proteomics of the same sample digests (Figure 1). Proteins are extracted, digested with trypsin and labelled. After pooling, a small aliquot is removed and fractionated using basic reverse-phase (bRP) chromatography with six non-enriched fractions collected. In the case of phospho proteomics (Phospho-SysQuant), in parallel the bulk of the analytical sample is processed using immobilised metal affinity chromatography (IMAC) to enrich phosphopeptides that are then fractionated using bRP and six enriched fractions collected. The resulting 12 fractions (6 x non-enriched, 6 x enriched) are analysed by LC-MS/MS on an Orbitrap Fusion Tribrid mass spectrometer with in-line uHPLC.
Using Phospho-SysQuant as an example we have typically quantified 7 to 10,000 unique proteins and 15 to 30,000 individual phosphorylation sites with the highest numbers seen in cultured human cells with tissues and samples from other species generally having lower coverage. The typical turnaround time for 1 x 18-plex experiment is 6 to 8 weeks.
Contact us for an example copy of our final analysis report.
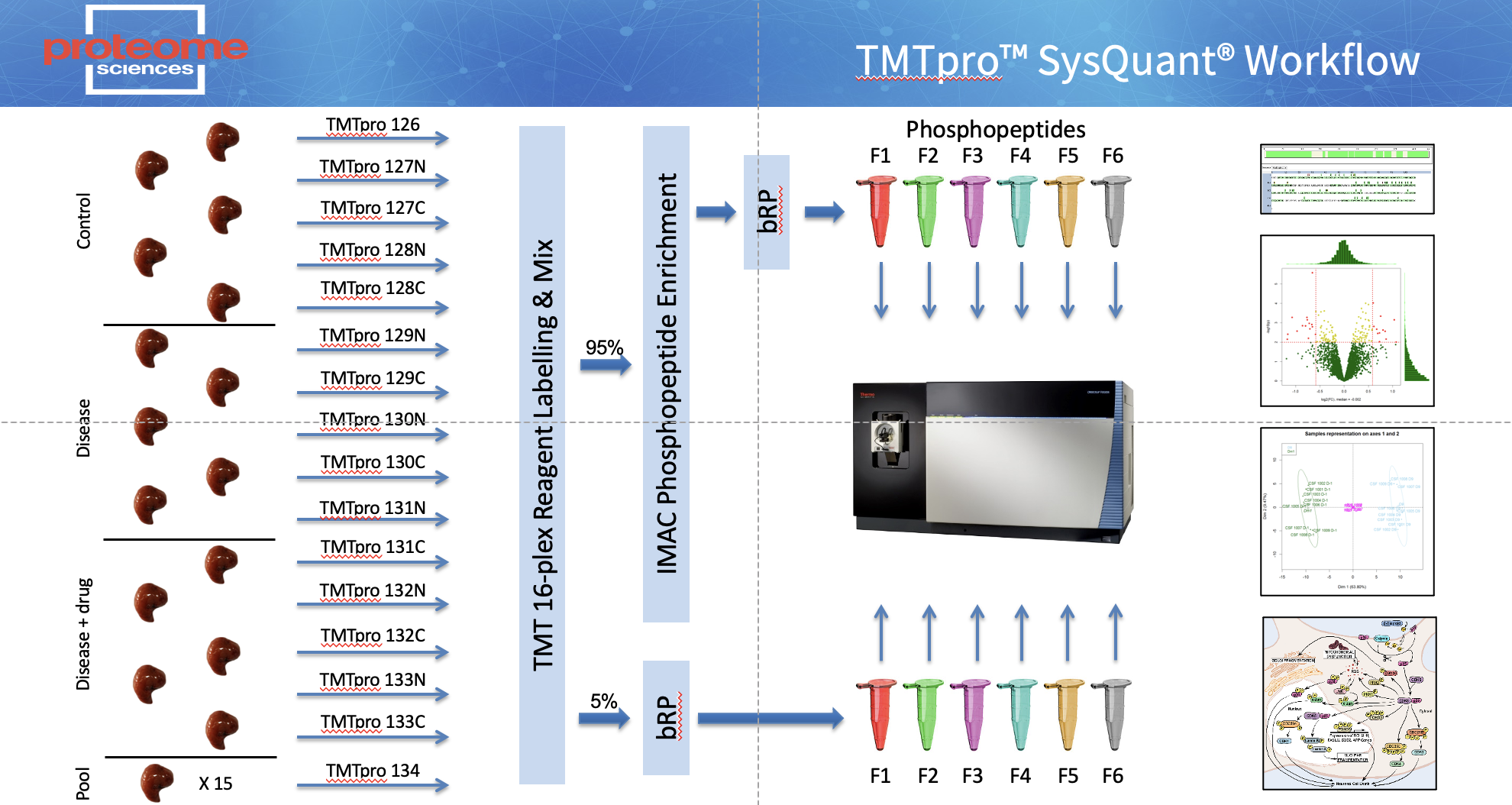
TMTpro based SysQuant Workflow
For each set of up to 11, 18 or 35 samples we generate 2 sets of TMT labelled both peptides and in this example phospho peptides samples. To sequence the protein backbone 5% of the sample peptides are analysed in a conventional LC-MS2 workflow to sequence the proteome. in the case of protein phosphorylation, the phospho-peptides in the remaining 95% of the sample are enriched via a metal affinity (IMAC) column and sequenced in a second LC-MS2 run.
Samples that can be analysed
- Primary tissues
- Cell culture
Typical Metrics (cell culture / tissue matrices reporting pSer, pThr, pTyr)
- 15,000 quantified sites
- 50,000 quantified peptides
- 500,000 quantitative measurements
SysQuant can be adapted for a variety of differing workflows to quantify a variety of PTM’s
Quantification of other PTM’s will employ a different enrichment stage, such as a IP pull down using a general specific antibody pull down targeted to the PTM of interest.
- Phospho-SysQuant - The proteome and phosphoproteome.
- Acetyl-SysQuant - The proteome and acetylproteome.
- Ubi-SysQuant - The proteome and ubiquitylome.
Other PTM analysis are available on request
- Methylation
- Nitrosylation
- Di-Sulphide bond localisation
- Glycosylation
Tissue and species used in Phospho-SysQuant
| Human Tissue | Other Species | Cell lines |
|---|---|---|
| Pancreas | Mouse Brain | Breast Cancer |
| Liver | Mouse PDX tumour | Pancreatic Cancer |
| Brain | Mouse PBMC | Melanoma |
| Muscle | Rat Brain | Colorectal Cancer |
| Skin | Rat Liver | iPSC |
| Adipose | Zebrafish | |
| PBMC | ||
| Lung |
An example report is available upon request.
References/Posters
1. Quantification of pancreatic cancer proteome and phosphorylome: indicates molecular events likely contributing to cancer and activity of drug targets - Dave Britton et al. Proteome Sciences authored paper describing the use of the first version of SysQuant to analyse protein and phosphorylation site changes in [pancreatic tumors relative to their healthy tissue backgrounds. Uncovered many sites and processes relating to epithelial to mesenchymal transformation.
“A multiomic approach to characterize the temporal sequence in Alzheimer's disease-related pathology” A new reference co-authored by Proteome Sciences and University Eastern Finland, coupling microarray-based transcriptomics and MS-based phospho-proteomics assay to determine mRNA, protein, and phosphopeptide (SysQuant) expression levels from 71 autopsied temporal cortical samples, with varying degree of AD-related neurofibrillary pathology. With computational analysis, they identified disease-related transcript, protein and phosphopeptide expression patterns, associated with distinct biological processes and cell types. Further observation of the obtained gene product expression patterns allowed for generation of a hypothetical temporal sequence of the changes in the activity of specific processes at different stages of AD-related neurofibrillary pathology.
For FAQ’s on Unbiased Protein Biomarker Discovery based Proteomics please follow this link.
| What’s included |
|
| Material required |
|
| Typical turnaround time |
|